A first-principles reassessment of the Fe-N phase diagram in the low-nitrogen limit
Journal of Alloys and Compounds
775, 758-768
2019
A1
This is a debugging block
This is a debugging block
This is a debugging block
This is a debugging block
This is a debugging block
This is a debugging block
The excellent mechanical properties of maraging steels are ascribed to nanometer-sized intermetallics which precipitate during aging in the ductile very low carbon Ni-martensite. Their wear and fatigue properties can be improved by nitriding. The non-equilibrium precipitation reactions in Fe-Ni-Co-Mo maraging steels are studied during an aging heat treatment executed in a nitriding atmosphere. The precipitates formed during the initial stages of precipitation are characterized with transmission electron microscopy and atom probe tomography. Spherical intermetallic precipitates having a diameter of around 3 nm were detected in the aged, bulk material. These ω-type precipitates formed during the early stages of aging, have a trigonal crystal lattice and their chemical composition is close to (Fe,Ni)7Mo2. In the nitrided layer, Mo-N disc-shaped nitrides on the {100} martensitic lattice having a diameter of 3 to 4 nm were found but their exact crystal structure could not be determined with microstructural characterization techniques. Density functional theory calculations confirmed that a single layer of Mo atoms, substituting Fe on the {100} plane of the Fe-matrix, is stable and showed that the N atoms prefer to be in the Mo-layer, on the octahedral sites with Fe as nearest neighbors.
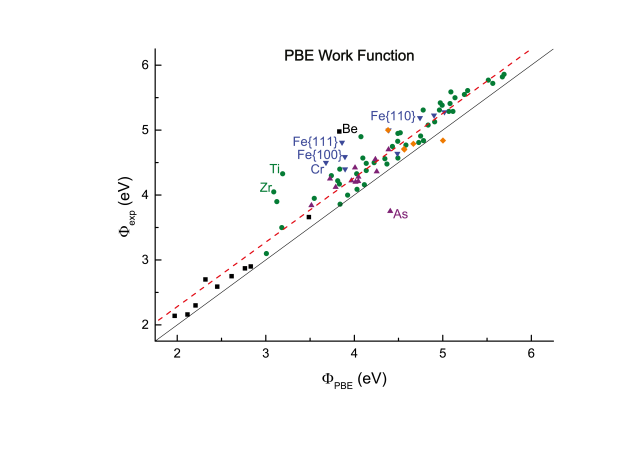
Density-functional theory (DFT) predictions of materials properties are becoming ever more widespread. With increased use comes the demand for estimates of the accuracy of DFT results. In view of the importance of reliable surface properties, this work calculates surface energies and work functions for a large and diverse test set of crystalline solids. They are compared to experimental values by performing a linear regression, which results in a measure of the predictable and material-specific error of the theoretical result. Two of the most prevalent functionals, the local density approximation (LDA) and the Perdew-Burke-Ernzerhof parametrization of the generalized gradient approximation (PBE-GGA), are evaluated and compared. Both LDA and GGA-PBE are found to yield accurate work functions with error bars below 0.3 eV, rivaling the experimental precision. LDA also provides satisfactory estimates for the surface energy with error bars smaller than 10%, but GGA-PBE significantly underestimates the surface energy for materials with a large correlation energy.
 Open Access version available at UGent repository
Open Access version available at UGent repositoryThis is a debugging block
This is a debugging block
This is a debugging block
This is a debugging block
This is a debugging block
This is a debugging block